
Abstract
A combined experimental and theoretical study on molecular and vibrational structure of E-N¢ (ICINH) had been carried out. The FTIR, FT-Raman and UV-Vis spectra of ICINH were recorded in the solid phase. The optimized geometry was calculated by B3LYP method with 6-311++G(d,p) level of basis set. The harmonic vibrational frequencies, IR intensities and Raman scattering activities of the title compound were calculated at same level of theory. The scaled theoretical wavenumber showed very good agreement with the experimental values. The mulliken charges and thermodynamic functions of the ICINH were also performed at same level of theory. NLO and a study on the electronic properties such as excitation energies and wavelength, were performed by TD-DFT approach. HOMO–LUMO energy gap was also calculated and interpreted.
Author Contributions
Academic Editor: Dr. Ashish Kumar, Associate Professor and HOD -Department of Chemistry, Lovely Professional University Phagwara
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 D Sumathi, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Indole is an aromatic heterocyclic organic compound with a bicyclic structure. It consist of a six-member benzene ring fused with five-member nitrogen containing pyrrole ring. It is of interest as it can be compared with tryptophane residue 1. The derivative of indole is present in both-animal and plants. The most important compound of this group is tryptophan, an essential amino acid in the human diet, which a 3-substituted indole 2. Another important indole derivative is the indole-3-acetic acid, a phytohormone, coordinating several growths processing of plants 3. The biological activity of the indole derivatives is in connection with the nature of substitution in position 3, on the pyrrole ring 4. Indole derivatives have an important role through individual biological functions. It is present in the side chain of amino acid tryptophan. The chemical and spectroscopic properties of indole derivatives have been subject of many experimental and theoretical investigations 5, 6, 7. The indole tryptamine if one of the biogenic monoamines would play anti-tumor effects by either inhibiting cancer cell proliferation or stimulating the anti-cancer immunity 8. DFT method has become an efficient tool in the prediction of molecular structure of organic molecules and in evaluating various molecular properties like conjugation, hydrogen bonding, vibrational frequencies and IR & Raman activities of the bioactive molecule 9, 10, 11, 12, 13.
The new Donor-π-Acceptor type dyes D1-3 carrying 3-(1-hexyl-1H-indol-3-yl)-2-(thiophen-2-yl) acrylonitrile as backbone with three different acceptor units were designed and synthesized by Babu, 14, using sensitizers for solar cell application. The new dyes were characterized by various spectral and elemental analyses. Their optical and electrochemical properties were investigated using spectrophotometry and cyclic voltammetry, respectively. The DFT study was carried out to investigate their Frontier MO energy states. The above study reveals that the dye carrying 4-aminobenzoic acid as an acceptor showed the highest photovoltaic efficiency among the three dyes. This can be attributed to the longer electron lifetime and lower recombination rates. Additionally, a single crystal XRD study confirms the structure of a key intermediate.
The stability of the syn and anti structures of the non-steroidal anti-inflammatory drug indomethacin were investigated by Hassan et al., 15, using the DFT/B3LYP and ab initio MP2 calculations with the 6-311G(d,p) basis set. The molecule indomethacin was predicted at the DFT and MP2 levels of calculation to have the syn (C1N7C10C18 ~ 40˚) form being about 1.7 and 1.5 kcal/mol, respectively, lower in energy than the anti (C1N7C10C18 ~ 140˚) structure. The calculated CNCC torsional angles for the chlorobenzene and indole rings syn-anti conformational interconversion was in a good qualitative agreement with the reported XRD angles (C1N7C10C18 ~ 29 and 155˚) for the syn and anti conformers, respectively. Indomethacin was estimated from the calculated Gibb’s free energies to have an equilibrium mixture of 95% syn and 5% anti structures at 298.15 K. The vibrational wavenumbers were calculated at the same level of theory. The complete vibrational assignments were provided on the basis of theoretical and NCA combined with experimental IR and Raman data of the molecule. The analysis of the observed spectra supported the presence of indomethacin in only one conformation at room temperature.
The (E)-2-(2-hydroxybenzylidenamino)-3-(1H-indol-3yl) propionic acid was synthesized by Saleem et al., 7. To identify the stable structure, the theoretical conformational analysis was performed. The optimized molecular bond parameters were calculated using B3LYP/6-31G(d,p) basis set. The hyperconjugative interaction energy (E(2)) and EDs of donor (i) and acceptor (j) bonds were calculated using NBO analysis. The dipole moment (µ) and first order hyperpolarizability (β0) were calculated. The band gap energy was analyzed by UV–Vis recorded spectra and compared with theoretical band gap (TD-DFT/B3LYP/6-31G(d,p)) value. The intra-molecular hydrogen bonding interaction was studied between nitrogen and hydroxyl hydrogen (N-H---O).
The molecular structure, complete vibrational spectra and the quantum mechanical calculations of the title compound were not yet reported. Therefore, the vibrational spectrum and the quantum mechanical calculations for the title compound in the ground state by DFT method with the standard 6-311++G(d,p) level of basis set were reported. Electronic absorption spectra of the title compound were predicted by using TD-DFT method. The excitation energies, wavelength and oscillator strengths were obtained at the same level of theory. Besides the molecular parameters, dipole moments, NLO, thermodynamic properties, linear polarizability and first hyperpolarizability, were calculated. The results obtained from theoretical calculations and experimental were compared.
Experimental Details
Synthesis Procedure
1H-indole-3-carbaldehyde (1.45 g, 0.01 mol) and isonicotinic acid hydrazide (1.37 g, 0.01 mol) were added to ethanol (10 ml) and stirred for an hour in the presence of hydrochloric acid to form a white precipitate. The precipitate was washed with sodium bicarbonate solution and filtered and again washed with petroleum ether (40–60%) and dried in air. The compound was recrystallized from absolute ethanol.
Results and Discussion
Molecular Geometry
The optimized structural parameters of E-N¢ are calculated using B3LYP/6-311++G(d,p) basis set and listed in Table 1. The optimized structure is shown in Figure 1. The title molecule consists of pyridine and indole ring fused by hydrazone linkage. In ICINH, the hydrazone linkage plays an important role. For the carbonyl (C21=O29) bond length in hydrazone link is calculated about 1.223Å using DFT and corresponding X-ray data value is 1.236Å. The bond length of N18-N19 is acted as bridge between the phenyl and pyridine ring and its bond length is calculated using DFT calculation at 1.368Å, while the corresponding X-rays value is 1.392Å. Similarly, the C21-N19/C16=N18 bond lengths are calculated as: 1.384 : DFT; 1.330Å : X-rays data/1.288 : DFT; 1.278Å : X-rays data, respectively. In the present study, the average value of C-C bond lengths in indole ring is 1.403Å (DFT). Bikas et al., 16, observed the bond angle of O-C-C at 120.6˚, which was in consistent with calculated value 121.49˚ (O29-C21-C22) and its corresponding DFT value is positively deviated (~ 1˚) from the calculated value. The dihedral angles are also calculated and listed in Table 1.
Figure 1.The optimized molecular structure of ICINH
| Bond Parameters | B3LYP/6-311++G(d,p) | XRDa |
| Bond Lengths (Å) | ||
| C1-C2 | 1.416 | |
| C1-C6 | 1.396 | |
| C1-N15 | 1.383 | 1.347 |
| C2-C3 | 1.404 | |
| C2-C8 | 1.451 | 1.445 |
| C3-C4 | 1.387 | |
| C3-H9 | 1.084 | |
| C4-C5 | 1.408 | |
| C4-H10 | 1.084 | |
| C5-C6 | 1.388 | |
| C5-H11 | 1.084 | |
| C6-H12 | 1.084 | |
| C7-C8 | 1.385 | |
| C7-H14 | 1.078 | |
| C7-N15 | 1.372 | |
| C8-C16 | 1.455 | 1.437 |
| H13-N15 | 1.007 | |
| C16-H17 | 1.087 | |
| C16-N18 | 1.288 | 1.278 |
| N18-N19 | 1.368 | 1.392 |
| N19-H20 | 1.019 | |
| N19-C21 | 1.384 | 1.33 |
| C21-C22 | 1.496 | 1.495 |
| C21-O29 | 1.223 | 1.236 |
| C22-C23 | 1.398 | |
| C22-C24 | 1.401 | |
| C23-C25 | 1.388 | |
| C23-H26 | 1.083 | |
| C24-H27 | 1.082 | |
| C24-N31 | 1.335 | |
| C25-C28 | 1.394 | |
| C25-H29 | 1.084 | |
| C28-H30 | 1.0867 | |
| C28-N31 | 1.336 | |
| Bond Angles (°) | ||
| C2-C1-C6 | 122.55 | |
| C2-C1-N15 | 107.12 | 109.56 |
| C6-C1-N15 | 130.34 | |
| C1-C2-C3 | 118.87 | |
| C1-C2-C8 | 107.31 | |
| C3-C2-C8 | 133.81 | |
| C2-C3-C4 | 118.9 | |
| C2-C3-H9 | 120.86 | |
| C4-C3-H9 | 120.24 | |
| C3-C4-C5 | 121.19 | |
| C3-C4-H10 | 119.6 | |
| C5-C4-H10 | 119.2 | |
| C4-C5-C6 | 121.18 | |
| C4-C5-H11 | 119.4 | |
| C6-C5-H11 | 119.42 | |
| C1-C6-C5 | 117.31 | |
| C1-C6-H12 | 121.52 | |
| C5-C6-H12 | 121.17 | |
| C8-C7-H14 | 130.27 | |
| C8-C7-N15 | 109.75 | |
| H14-C7-N15 | 119.85 | 123.3 |
| C2-C8-C7 | 106.01 | |
| C2-C8-C16 | 123.89 | 130.5 |
| C7-C8-C16 | 129.95 | |
| C1-N15-C7 | 109.81 | |
| C1-N15-H13 | 125.49 | |
| C7-N15-H13 | 124.69 | |
| C8-C16-H17 | 115.93 | |
| C8-C16-N18 | 130.93 | |
| H17-C16-N18 | 113.14 | |
| C16-N18-N19 | 117.96 | 113.29 |
| N18-N19-H20 | 119.23 | |
| N18-N19-C21 | 123.39 | 120.33 |
| H20-N19-C21 | 112.73 | |
| N19-C21-C22 | 119.89 | 114.64 |
| N19-C21-O29 | 118.6 | 124.7 |
| C22-C21-O29 | 121.49 | 120.6 |
| C21-C22-C23 | 117.13 | |
| C21-C22-C24 | 124.87 | |
| C23-C22-C24 | 117.85 | |
| C22-C23-C25 | 119.05 | |
| C22-C23-H26 | 119.22 | |
| C25-C23-H26 | 121.73 | |
| C22-C24-H27 | 120.51 | |
| C22-C24-N31 | 123.46 | |
| H27-C24-N31 | 116.03 | |
| C23-C25-C28 | 118.39 | |
| C23-C25-H29 | 121.19 | |
| C28-C25-H29 | 120.41 | |
| C25-C28-H30 | 120.46 | |
| C25-C28-N31 | 123.5 | |
| H30-C28-N31 | 116.05 | |
| C24-N31-C28 | 117.73 | |
| C6-C1-C2-C3 | -0.05 | |
| C6-C1-C2-C8 | -179.54 | |
| N15-C1-C2-C3 | 179.79 | |
| N15-C1-C2-C8 | 0.29 | |
| C2-C1-C6-C5 | -0.29 | |
| C2-C1-C6-H12 | 179.72 | |
| N15-C1-C6-C5 | 179.92 | |
| N15-C1-C6-H12 | -0.07 | |
| C2-C1-N15-C7 | -0.26 | |
| C2-C1-N15-H13 | 179.33 | |
| C6-C1-N15-C7 | 179.56 | |
| C6-C1-N15-H13 | -0.85 | |
| C1-C2-C3-C4 | 0.41 | |
| C1-C2-C3-H9 | -179.24 | |
| C8-C2-C3-C4 | 179.74 | |
| C8-C2-C3-H9 | 0.09 | |
| C1-C2-C8-C7 | -0.22 | |
| C1-C2-C8-C16 | -176.16 | |
| C3-C2-C8-C7 | -179.61 | |
| C3-C2-C8-C16 | 4.45 | |
| C2-C3-C4-C5 | -0.44 | |
| C2-C3-C4-H10 | 179.88 | |
| H9-C3-C4-C5 | 179.21 | |
| H9-C3-C4-H10 | -0.47 | |
| C3-C4-C5-C6 | 0.11 | |
| C3-C4-C5-H11 | -179.77 | |
| H10-C4-C5-C6 | 179.78 | |
| H10-C4-C5-H11 | -0.09 | |
| C4-C5-C6-C1 | 0.26 | |
| C4-C5-C6-H12 | -179.75 | |
| H11-C5-C6-C1 | -179.87 | |
| H11-C5-C6-H12 | 0.12 | |
| H14-C7-C8-C2 | -175.75 | |
| H14-C7-C8-C16 | -0.14 | |
| N15-C7-C8-C2 | 0.06 | |
| N15-C7-C8-C16 | 175.67 | |
| C8-C7-N15-C1 | 0.12 | |
| C8-C7-N15-H13 | -179.48 | |
| H14-C7-N15-C1 | 176.44 | |
| H14-C7-N15-H13 | -3.16 | |
| C2-C8-C16-H17 | 20.44 | |
| C2-C8-C16-N18 | -158.81 | |
| C8-C16-N18-N19 | -154.47 | |
| H17-C16-N18-N19 | 26.29 | |
| C16-N18-N19-H20 | 2.17 | |
| C16-N18-N19-C21 | -177.1 | |
| N18-N19-C21-C22 | 21.38 | |
| N18-N19-C21-O32 | 175.42 | |
| H20-N19-C21-C22 | 21.29 | |
| H20-N19-C21-O32 | -160.76 | |
| N19-N21-C22-C23 | 176.81 | |
| N19-N21-C22-C24 | -5.23 | |
| O32-C21-C22-C23 | -156.35 | |
| O32-C21-C22-C24 | 28.1 | |
| C21-C22-C23-C25 | 25.76 | |
| C21-C22-C23-H26 | -149.79 | |
| C24-C22-C23-C25 | -177.41 | |
| C24-C22-C23-H26 | 2.29 | |
| C21-C22-C24-H27 | -1.54 | |
| C21-C22-C24-N31 | 178.16 | |
| C23-C22-C24-H27 | -3.35 | |
| C23-C22-C24-N31 | 175.99 | |
| C22-C23-C25-C28 | -178.87 | |
| C22-C23-C25-H29 | 0.47 | |
| H26-C23-C25-C28 | 1.25 | |
| H26-C23-C25-H29 | -179.2 | |
| C22-C24-N31-C28 | -178.45 | |
| H27-C24-N31-C28 | 1.1 | |
| C23-C25-C28-H30 | 0.92 | |
| C23-C25-C28-N31 | -179.72 | |
| H29-C25-C28-H30 | 179.8 | |
| H29-C25-C28-N31 | 0.18 | |
| C25-C28-N31-C24 | 0.25 | |
| H30-C28-N31-C24 | -179.37 |
Vibrational Assignments
The harmonic vibrational frequencies were calculated using B3LYP/6-311++G(d,p) basis set. The title molecule belongs to C1 point group symmetry and it had 90 vibrational normal modes of same symmetry species are listed in Table 2.1. The internal and symmetry coordinates of ICINH are listed in Table 2.1 and Table 2.2, respectively. The percentage of PED obtained from MOLVIB program package was used for assigning vibrational peaks. The exclusion of anharmonicity factor and the level of basis set used, certain theoretical frequencies are not matched with that of the experimental values. Hence linear scaling procedure is adopted to scale down the frequency values. In this study, we have followed scaling factor of 0.968 for DFT 17. The observed FT-IR, FT-Raman and simulated spectra of ICINH are shown in Figure 2 and Figure 3, respectively.
Table 2. 1. Possible internal co-ordinates of ICINH| S.No | Type | Fragment type | Definition |
| 8-Jan | C-C | Double ring | C1-C2, C2-C3, C3-C4, C4-C5, C5-C6, C6-C1, C2-C8, C7-C8 |
| 9,10 | C-N | C1-N15, C7-N15 | |
| 15-Nov | C-H | C3-H9, C4-H10, C5-H11, C6-H12, C7-H14 | |
| 16 | N-H | N15-H13 | |
| 17-20 | C-C | Pyridine ring | C22-C23, C22-C24, C23-C25, C25-C28 |
| 21,22 | C-N | C24-N31, C28-N31 | |
| 23-26 | C-H | C23-H26, C24-H27, C25-H29, C28-H30 | |
| 27,28 | C-C | Out-of the ring | C8-C16, C21-C22 |
| 29 | C=O | C21-O32 | |
| 30 | N-H | N19-H20 | |
| 31 | C-H | C16-H17 | |
| 32,33 | C-N | C16-N18, C21-N19 | |
| 34 | N-N | N18-N19 | |
| In-plane bending | |||
| 35-45 | C-C(N)-C(N) | Double ring | C1-C2 -C3, C2-C3-C4, C3-C4 -C5, C4-C5 -C6, C5-C6 -C1, C6-C1-C2, C1-C2-C8, C2-C8-C7, C8-C7-N15, C7-N15-C1 |
| N15-C1 -C2 | |||
| 46-55 | C(N)-C-H | C2-C3 -H9, C4-C3 -H9, C3-C4 -H10, C5-C4 -H10, C4-C5 -H11, C6-C5 -H11, C1-C6 -H12, C5-C6 -H12, C8-C7 -H14, N15-C7 -H14 | |
| 56,57 | C-N-H | C1-N15-H13, C7-N15-H13 | |
| 58,59 | C-C-C(N) | C6-C1-N15, C3-C2-C8 | |
| 60,61 | C-C-H | Out-of the ring | C8-C16-H17, N18-C16-H17 |
| 62,63 | C-C-N | C8-C16-N18, C22-C21-N19 | |
| 64,65 | C-N-N | C16-N18-N19, C21-N19-N18 | |
| 66,67 | C(N)-N-H | C21-N19-H20, N18-N19-H20 | |
| 68,69 | C(N)-C=O | C22-C21-O32, N19-C21-O32 | |
| 70-73 | C-C-C | C21-C22 -C23, C21-C22-C24, C2-C8 -C16, C7-C8-C16 | |
| 74-79 | C-C(N)-C(N) | Pyridine ring | C22-C23 -C25, C23-C25-C28, C25-C28 -N31, C28-N31-C24, N31-C24-C22, C24-C22-C23 |
| 80-87 | C(N)-C-H | C22-C23-H26, C25-C23-H26, C22-C24-H27, N31-C24-H27, C23-C25-H29, C28-C25-H29, C25-C28-H30, N31-C28-H30 | |
| Out-of-plane bending | |||
| 88-98 | C(N)-C(N)-C(N)-C(N) | Double ring | C1-C2-C3-C4, C2-C3-C4-C5, C3-C4-C5-C6, C4-C5 -C6-C1, C5-C6-C1-C2, C6-C1-C2-C3, C1-C2 -C8-C7, C2-C8-C7-N15, C8-C7-N15-C1, C7-N15-C1-C2, N15-C1 -C2-C8 |
| 99-103 | H-C(N)-C-C(N) | H9-C3-C2-C4, H10-C4-C3-C5, H11-C5-C4-C6, H12-C6-C1-C5, H13-N15-C1-C7, H14-C7-C8-N15 | |
| 105,106 | C-C-C-C | Out-of the ring | C16-C8-C2-C7, C21-C22-C23-C24 |
| 107 | O-C-C-N | O32-C21-C22-N19 | |
| 108 | H-N-N-C | H20-N19-N18-C21 | |
| 109 | H-C-C-N | H17-C16-C8-N18 | |
| 110 | C-N-N-C | C16-N18-N19-C21 | |
| 111-114 | N-C-C-C | N19-C21-C22-C23, N19-C21-C22-C24, | |
| C2- C8- C16-N18, C7-C8-C16-N18 | |||
| 115-120 | C(N)-C(N)-C(N)-C(N) | Pyridine | C22-C23-C25-C28, C23-C25-C28-N31, C25-C28-N31-C24, C28-N31-C24-C22, N31-C24-C22-C23, C24-C22-C23-C25 |
| 121-124 | H-C-C-C | H26-C23-C22-C25, H27-C24-C22-N31, H29-C25-C23-C28, H30-C28-C25-N31 | |
| 125,126 | C(N)-C-C-C | Torsion | C3-C2-C8-C16, N15-C7-C8-C16 |
| 127,128 | C-C-C-C(N) | Butterfly | C8-C2-C1-C6, C3-C2-C1-N15 |
| Mode No | Exp. IR | Exp. Raman | Frequencies | Scaled | Red. Masses | Force | IR | Raman Intensity | Vibrational Assignments |
| frequencies | constants | Intensity | |||||||
| 1 | - | - | 23 | 22 | 5.6685 | 0.0017 | 0.05 | 41.46 | gring (90) |
| 2 | - | - | 31 | 30 | 6.2039 | 0.0035 | 0.26 | 47.75 | gring (93) |
| 3 | - | - | 38 | 37 | 4.7887 | 0.0041 | 0.29 | 50.62 | g(py)ring (97) |
| 4 | - | - | 73 | 70 | 6.1906 | 0.0192 | 0.52 | 0.88 | g(py)ring (98) |
| 5 | - | 89 | 93 | 90 | 6.2748 | 0.0318 | 0.06 | 10.04 | gring (89) |
| 6 | - | - | 132 | 128 | 6.7417 | 0.0692 | 0.31 | 3.54 | gC=O (69), gring (23) |
| 7 | - | - | 151 | 146 | 5.367 | 0.0716 | 0.35 | 3.71 | gring (66), gN−N (26) |
| 8 | - | - | 171 | 166 | 4.8316 | 0.0835 | 0.2 | 5 | gN−N(54), gring (37) |
| 9 | - | - | 196 | 190 | 5.1729 | 0.1176 | 0.19 | 3.45 | g(py)ring (67), Butterfly (21) |
| 10 | - | - | 223 | 216 | 4.1358 | 0.1213 | 2.07 | 1.51 | Butterfly (89) |
| 11 | - | - | 236 | 229 | 5.5815 | 0.1835 | 2.48 | 4.94 | β(py)ring (78) |
| 12 | - | - | 295 | 285 | 7.0353 | 0.3595 | 0.36 | 0.27 | tring (94) |
| 13 | - | 376 | 363 | 5.8348 | 0.4847 | 0.8 | 0.48 | gring (62), goutC−C(21) | |
| 14 | - | - | 402 | 389 | 3.5462 | 0.3379 | 0.58 | 1.55 | goutC−N(43), gN−N(18), gring(14) |
| 15 | - | - | 418 | 405 | 5.2085 | 0.5367 | 0.63 | 1.35 | gring (69), g(py)ring (24) |
| 16 | - | - | 426 | 412 | 2.5799 | 0.2753 | 6.89 | 0.44 | goutC−C(50), bC−N(22), bN−H(10) |
| 17 | 426 | - | 440 | 426 | 1.5194 | 0.1731 | 7.82 | 0.87 | gN−H(78) |
| 18 | - | - | 449 | 434 | 3.4966 | 0.415 | 3.38 | 0.38 | goutC−C(56), gC−H (22) |
| 19 | - | - | 498 | 482 | 5.8526 | 0.8561 | 0.62 | 1.6 | βring (49),goutC−C(19), gring (18) |
| 20 | - | - | 552 | 535 | 5.1322 | 0.9221 | 1.85 | 2.76 | gC=N(61), gring (26) |
| 21 | - | - | 563 | 545 | 5.496 | 1.0281 | 0.54 | 2.03 | βring (72) |
| 22 | - | - | 582 | 564 | 2.8828 | 0.5762 | 1.76 | 4.01 | βoutC-C (46), βN‒H (23) |
| 23 | - | - | 589 | 570 | 3.2755 | 0.6698 | 7.32 | 2.24 | goutN−H(63), βC−H (18) |
| 24 | 581 | - | 606 | 587 | 5.2868 | 1.1458 | 11.97 | 3.94 | β(py)ring (68), goutN−H(22) |
| 25 | 615 | - | 637 | 616 | 6.325 | 1.5108 | 2.52 | 1.88 | βoutC-C (58), βC‒H (20), βN‒H (14) |
| 26 | - | - | 643 | 623 | 2.7471 | 0.6694 | 0.95 | 1.01 | βring (52), gC−H (24) |
| 27 | - | - | 668 | 647 | 2.6474 | 0.6964 | 2.08 | 15.49 | βN-N (50), βC‒N (18), β(py)ring (14) |
| 28 | 682 | - | 702 | 679 | 4.4972 | 1.3055 | 1.36 | 2.28 | βring (51), β(py)ring (27) |
| 29 | - | - | 719 | 696 | 2.1818 | 0.6637 | 3.21 | 0.31 | βoutC-C (47), βoutC−H (20), βring (17) |
| 30 | - | - | 748 | 724 | 3.0533 | 1.0053 | 12.23 | 4.31 | βoutC-N (57), βpyring (18), βC=O (13) |
| 31 | - | - | 751 | 727 | 1.4353 | 0.477 | 18.39 | 0.67 | gC-H (79) |
| 32 | - | - | 754 | 730 | 4.3578 | 1.4603 | 3.85 | 4.77 | gC-H (74), β(py)ring (13) |
| 33 | 752 | - | 774 | 749 | 3.9285 | 1.3867 | 0.53 | 1.49 | βoutC-H (48), βring (21) |
| 34 | - | - | 784 | 759 | 4.6888 | 1.6972 | 4.98 | 9.52 | βring (56), β(py)ring (23) |
| 35 | 793 | 795 | 828 | 802 | 1.3744 | 0.5555 | 1.89 | 1.51 | gC-H (88) |
| 36 | - | - | 840 | 813 | 1.9452 | 0.808 | 2.26 | 1.91 | g(py)C-H (92) |
| 37 | 830 | - | 855 | 828 | 1.4952 | 0.6446 | 0.98 | 0.25 | βC=O (60), β(py)ring (19) |
| 38 | - | - | 877 | 849 | 3.9613 | 1.796 | 23.55 | 3.59 | βC=N (45), βring (24), β(py)ring (18) |
| 39 | - | - | 893 | 864 | 4.4426 | 2.0861 | 3.75 | 0.23 | βring (78) |
| 40 | - | - | 919 | 890 | 1.4668 | 0.7301 | 1.25 | 6.07 | gC-H (91) |
| 41 | 912 | - | 944 | 913 | 1.3666 | 0.717 | 0.23 | 0.11 | gC-H (87) |
| 42 | - | - | 949 | 919 | 1.3956 | 0.7404 | 0.86 | 0.11 | g(py)C-H (90) |
| 43 | - | - | 981 | 949 | 1.2808 | 0.7258 | 0.01 | 0.06 | bN-H (59), bC−H(21) |
| 44 | 953 | - | 986 | 955 | 1.4487 | 0.8303 | 0.17 | 0.01 | g(py)C-H (92) |
| 45 | - | - | 1006 | 974 | 1.3774 | 0.8218 | 0.47 | 0.09 | g(py)C-H (92) |
| 46 | - | - | 1035 | 1002 | 2.1293 | 1.3435 | 1.87 | 6.9 | υC-C (41), βC‒H (28) |
| 47 | 1006 | 1009 | 1040 | 1007 | 4.2574 | 2.715 | 2.2 | 2.57 | υ(py)C-C (52), β(py)C‒H (29) |
| 48 | - | - | 1059 | 1025 | 3.2592 | 2.1539 | 0.09 | 13.5 | υ(py)C-C (68), β(py)C‒H (24) |
| 49 | 1047 | 1041 | 1066 | 1032 | 4.5472 | 3.0425 | 4.45 | 1.84 | υoutC-C (45), βring (20), βC‒H (14) |
| 50 | - | - | 1109 | 1074 | 3.4326 | 2.4881 | 18.31 | 16.64 | υN-N(52), β(py)C‒H (18), βoutC‒H (13) |
| 51 | - | 1092 | 1129 | 1093 | 1.5981 | 1.2012 | 5.03 | 1.73 | β C-H (47), βN‒H (22), υC‒N(17) |
| 52 | - | - | 1139 | 1102 | 1.4616 | 1.1167 | 0.86 | 0.43 | β(py)C-H (50), υ(py)C‒C (27) |
| 53 | - | - | 1155 | 1118 | 1.4763 | 1.1596 | 8.68 | 0.69 | βC-H (69) |
| 54 | 1126 | 1125 | 1160 | 1123 | 2.4869 | 1.9719 | 14.1 | 1.24 | υoutC-C (42), υoutC‒N (28) β(py)C‒H (15), |
| 55 | - | - | 1178 | 1140 | 1.1543 | 0.9431 | 0.36 | 0.39 | υC-C (53), υC‒N (19), βC‒H (15) |
| 56 | - | - | 1225 | 1186 | 1.4787 | 1.3081 | 2.73 | 1.51 | β(py)C-H (72), υ(py)C‒N (20) |
| 57 | - | - | 1251 | 1211 | 2.7583 | 2.5422 | 12.8 | 0.79 | βC-H (52), υC‒N(18), βN‒H (14) |
| 58 | - | - | 1263 | 1222 | 1.8362 | 1.7252 | 7.35 | 10.89 | υC-N (54), βC‒H (31) |
| 59 | 1244 | 1254 | 1285 | 1244 | 7.2124 | 7.0219 | 2.07 | 1.25 | υ(py)C-N (59), β(py)C‒H (22) |
| 60 | - | 1284 | 1325 | 1282 | 2.4286 | 2.5111 | 0.37 | 5.82 | υC-C (62), βC‒H (20) |
| 61 | 1298 | - | 1339 | 1296 | 2.3836 | 2.5178 | 54.46 | 0.89 | β C‒H (49), υC‒N(19), βoutC‒H (17) |
| 62 | - | - | 1361 | 1318 | 3.0228 | 3.3 | 71.69 | 18.1 | υ(py) C‒C (50), βC‒H (21), υoutC‒C (12) |
| 63 | - | - | 1365 | 1322 | 1.2876 | 1.4144 | 0.5 | 0.8 | υoutC‒N (58), βC‒H (23) |
| 64 | 1336 | 1345 | 1371 | 1327 | 5.9533 | 6.5908 | 6.32 | 4.61 | υC‒C (72), βC‒H (21) |
| 65 | 1360 | 1368 | 1410 | 1364 | 1.5509 | 1.8154 | 16.15 | 8.86 | boutC‒H (59), υC‒N (15), βN‒H (12) |
| 66 | - | - | 1442 | 1396 | 2.1721 | 2.6626 | 6.54 | 4.66 | υC‒C (49), βC‒H (27) |
| 67 | 1408 | - | 1447 | 1401 | 2.2447 | 2.7699 | 2.37 | 1.22 | β(py)C‒H (65), υ(py) (24) |
| 68 | - | - | 1473 | 1426 | 1.7655 | 2.2568 | 30.11 | 9.65 | βoutN-H (60), υoutC-N (17), υoutC‒C (14) |
| 69 | - | - | 1483 | 1436 | 2.3645 | 3.064 | 4.89 | 0.81 | βC-H (54), υC-C (21), βoutN‒H (18) |
| 70 | 1447 | 1455 | 1508 | 1460 | 2.2353 | 2.9957 | 1.61 | 3.14 | β(py) C-H (48), υ(py) (23), υoutC-C (10) |
| 71 | - | - | 1524 | 1475 | 2.7967 | 3.8252 | 0.94 | 0.21 | υC−N (43), υC−C (24), βC−H (18) |
| 72 | 1501 | - | 1554 | 1505 | 4.4312 | 6.307 | 20.77 | 41.94 | υC=C (68), βC−H (19) |
| 73 | - | - | 1604 | 1553 | 5.2119 | 7.9032 | 1.13 | 0.34 | υ(py) C−C (69), βC−H (22) |
| 74 | - | 1563 | 1615 | 1563 | 5.7905 | 8.8973 | 0.09 | 6.24 | υC−C (53), βN−H (25) |
| 75 | - | - | 1628 | 1575 | 5.6711 | 8.851 | 11.33 | 13.99 | υ(py) C−N (56),υ(py)C−C (23),β(py)C−H(14) |
| 76 | - | - | 1657 | 1604 | 7.6432 | 12.3614 | 5.91 | 100 | υout.C=N (56), υC−C (23), βC−H (14) |
| 77 | 1608 | 1606 | 1658 | 1605 | 6.6961 | 10.8407 | 2.95 | 28.6 | υC−C (52), υC=N (28), βC−H (12) |
| 78 | 1677 | - | 1718 | 1663 | 7.9762 | 13.8766 | 100 | 14.72 | υC=O (71), βN−H (23) |
| 79 | - | - | 3147 | 3046 | 1.0879 | 6.3482 | 1.02 | 0.82 | υout.C−H (98) |
| 80 | - | - | 3150 | 3049 | 1.0892 | 6.366 | 4.06 | 2.6 | υas(py) C−H (95) |
| 81 | - | - | 3167 | 3066 | 1.0858 | 6.4172 | 0.15 | 0.55 | υasC−H (96) |
| 82 | 3078 | - | 3174 | 3073 | 1.0887 | 6.463 | 0.72 | 2.26 | υasC−H (96) |
| 83 | - | - | 3184 | 3082 | 1.0936 | 6.532 | 4.39 | 1.24 | υasC−H (95) |
| 84 | - | - | 3185 | 3083 | 1.0909 | 6.5186 | 2.94 | 2.8 | υas(py)C−H (92) |
| 85 | - | - | 3194 | 3092 | 1.0971 | 6.5933 | 3.03 | 5.67 | υsC−H (97) |
| 86 | - | - | 3201 | 3099 | 1.092 | 6.593 | 2.66 | 0.24 | υas(py)C−H (91) |
| 87 | 3107 | 3109 | 3202 | 3100 | 1.0948 | 6.6134 | 0.13 | 3.81 | υas(py)C−H (94) |
| 88 | 3159 | - | 3253 | 3149 | 1.097 | 6.8395 | 0.27 | 0.6 | υC−H (99) |
| 89 | - | - | 3471 | 3360 | 1.0748 | 7.6289 | 7.5 | 3.16 | υout.N−H (99) |
| 90 | 3531 | - | 3664 | 3540 | 1.0805 | 8.5473 | 21.55 | 1.77 | υN−H (100) |
Figure 2.The combined theoretical and experimental FT-IR spectra of ICINH
Figure 3.The combined theoretical and experimental FT-Raman spectra of ICINH
Ring Vibrations
The ring stretching vibrations are very prominent in the spectrum of pyridine and its derivatives and are highly characteristics of aromatic ring itself 18. The C−C stretching vibrations of pyridine derivatives usually appear in the region between 1650–1400 cm−1 and 1100−1000 cm−1 19. In the present study, the peaks identified at 1006 cm−1 in FT-IR and 1009 cm−1 in FT-Raman are assigned to C−C stretching vibrations. Due to the absence of peaks, the theoretically scaled values at 1553, 1318 and 1025 cm−1 are assigned as C−C stretching vibrations of the rest of other modes in pyridine ring .The presence of nitrogen in the ring of the pyridine structure gives rise to two C−N stretching vibrations. Identifying these vibrations is rather a difficult task as their vibrational frequency lies within the C−C stretching region. As expected, the peaks for C−N stretching vibrations are found at 1244 cm−1 in FT-IR and 1254 cm−1 in FT-Raman spectrum. The theoretically scaled values of this mode are found to be in good agreement with experimental values.
According to PED results, the C−C stretching vibrations of six and five members are assigned. The C−C stretching vibrations are observed at 1608, 1501 and 1336 cm−1 in FT-IR and 1606, 1563, 1345 and 1284 cm−1 in FT-Raman spectrum. The theoretically predicted scaled values are in excellent correlation with that of the experimental values.
C−H Vibrations
Four C−H bonds in the pyridine ring of the title molecule give rise to four C−H stretching vibrations. The hetero-aromatic structure shows the presence of C−H stretching vibrations in the region 3000−3100 cm−1 20, which is the characteristic region for ready identification of this structure. In this region, the bands are not affected appreciably by the nature of the substituents. In the present study, the peak at 3107 cm−1 in FT-IR and at 3109 cm−1 in FT-Raman are assigned to C−H stretching vibrations of the pyridine ring. The percentage of PED results show that these modes are very pure modes. Similarly, the peaks appeared at 3159 and 3078 cm−1 in FT-IR are ascribed to C−H stretching vibrations of the benzene ring of the title molecule.
C=O Vibrations
The characteristic IR absorption wavenumber of C=O is normally strong in intensity and found in the region 1600–1800 cm−1 21, 22. In the present study, the strong peak at 1677 cm−1 in FT-IR is assigned to C=O stretching. The calculated value of C=O stretching mode at B3LYP/6-311++G(d,p) shows good agreement with the experimental value. The C=O in-plane bending vibration is identified at 830 cm−1 in FT-IR. A peak for C=O out-of-plane bending vibration is not active in both IR and Raman spectra. Hence the theoretically predicted value (128 cm-1/mode no: 6) is assigned to this mode.
N−H Vibrations
In primary amines, usually the N–H stretching vibrations occur in the region 3600–3300 cm−1 23. In the present study, the N–H bond in the five member ring produce a well defined peak at 3531 cm−1 in FT-IR. A peak for the N19−H20 bond present at the out-of-the ring is not active in both IR and Raman. Hence, the theoretical scaled value of 3540 cm−1 is assigned to N15−H13 stretching mode. All the vibrations in this group are in excellent agreement with experimental results as well as with literature 24.
C=N and N−N Vibrations
The identification of C=N stretching vibrations are difficult task since these are usually coupled with ring stretching and C-H in-plane bending vibrations. A bond C21=N19 at out of the ring possesses three vibrational normal modes. Since all these vibrations are inactive in both the spectra, the theoretically predicted wavenumbers 1604, 849 and 535 cm−1 are ascribed to C=N stretching, in-plane bending and out-of-plane bending, respectively. Similarly, the N−N stretching and bending vibrations are not present in both IR and Raman. Hence, the theoretically scaled values of 1074, 647 and 166 cm−1 are attributed to N−N stretching, in-plane bending and out-of-plane bending vibrations, respectively.
NLO Property
Non linear effect arise from the interactions of electromagnetic fields in various media to produce new fields altered in phase, frequency, amplitude or other propagation characteristics from incident fields 25. The first hyperpolarizability (β0), dipole moment μ and polarizability α are calculated using DFT/6-311++G(d,p) basis set. The computed total static dipole moment (μ), the mean polarizability (α0) the mean first hyperpolarizability (β0), for the molecule under study are presented in Table 3 shows that the first order hyperpolarizability value play an important role in determining the NLO activity of the molecule. The first order hyper polarizability (β0) of the present molecule is 8.6424x10-30 esu while that of urea is 0.3728x10-30 esu. The (βo) of ICINH is 23 times greater than that of urea, hence, the molecule can be said to be highly NLO activity; which is naturally due to the contribution of oxygen atom which makes one part of molecule highly negative and other part as equally positive.
Table 3. The NLO measurements of ICINH| Parameters | B3LYP/6-311++G(d,p) |
| Dipole moment ( μ ) Debye | |
| μx | 1.6403 |
| μy | 0.4934 |
| μz | 0.1242 |
| Μ | 1.7174Debye |
| Polarizability ( α0 ) x10-30esu | |
| αxx | 351.00 |
| αxy | -4.96 |
| αyy | 194.94 |
| αxz | 1.53 |
| αyz | 4.90 |
| αzz | 132.91 |
| αo | 0.5902x10-30esu |
| Hyperpolarizability ( β0 ) x10-30esu | |
| βxxx | -1163.89 |
| βxxy | 31.69 |
| βxyy | 148.04 |
| βyyy | -106.30 |
| βxxz | -196.67 |
| βxyz | -19.08 |
| βyyz | 12.83 |
| βxzz | 46.85 |
| βyzz | -42.47 |
| βzzz | -35.40 |
| β0 | 8.6424x10-30esu |
NBO Analysis
The NBO analysis is performed on ICINH using B3LYP/6-311++G(d,p) basis set and are listed in Table 4. In the study, the π bonds have higher ED than the σ bonds. Due to this reason, the σ-σ* transitions have minimum delocalization energy than the π-π* transitions. It is evident from the Table 4. The ED of π(C30-N32) bond transfer energy 116.4, 52.38 kJ/mol to the acceptor orbitals: C22-C24 and C23-C25, respectively. There occurs a strong intra-molecular hyperconjugative interaction of π electron from C23-C25 bond to the π*C22-C24→C30-N32 bonds, which increases the EDs: 0.3281 and 0.3727 kJ/mol, respectively. The lone pair electrons are readily available for the interaction with excited electrons of antibonding orbital. During n-π* transition, more energy delocalization takes place: N19→C16-N18; C21→O29 and N15→C1-C2; C7-C8. The corresponding excitation energy values are, 102.42, 170.41 and 140.71, 160.41, respectively. In which, these (N19→C21-O29 & N15→C7-C8) two transitions give the strongest stabilization to the system.
Table 4. The Second order perturbation theory analysis of Fock Matrix in NBO basis for ICINH| Type | Donor NBO (i) | ED/e | Acceptor NBO (j) | ED/e | aE(2) | bE(j)-E(i) | cF(i,j) |
| KJ/mol | a.u. | a.u. | |||||
| σ -σ* | BD ( 1) C 1 - C 2 | 1.959 | BD*(1) C 1 - C 6 | 0.021 | 18.95 | 1.23 | 0.07 |
| BD*(1) C 2 - C 3 | 0.022 | 14.06 | 1.24 | 0.06 | |||
| BD*(1) C 2 - C 8 | 0.027 | 8.2 | 1.16 | 0.04 | |||
| BD*(1) C 3 - H 9 | 0.014 | 10.59 | 1.11 | 0.05 | |||
| BD*(1) C 6 - H 12 | 0.013 | 9.41 | 1.1 | 0.05 | |||
| BD*(1) C 8 - C 16 | 0.036 | 17.95 | 1.15 | 0.06 | |||
| BD*(1) H 13 - N 15 | 0.016 | 16.82 | 1.04 | 0.06 | |||
| π -π* | BD ( 2) C 1 - C 2 | 1.596 | BD*(2) C 3 - C 4 | 0.301 | 79.16 | 0.29 | 0.07 |
| BD*(2) C 5 - C 6 | 0.319 | 76.11 | 0.28 | 0.07 | |||
| BD*(2) C 7 - C 8 | 0.349 | 78.7 | 0.26 | 0.06 | |||
| σ -σ* | BD ( 1) C 1 - C 6 | 1.976 | BD*(1) C 1 - C 2 | 0.027 | 20.21 | 1.25 | 0.07 |
| BD*(1) C 1 - N 15 | 0.027 | 8.91 | 1.15 | 0.04 | |||
| BD*(1) C 2 - C 8 | 0.027 | 5.86 | 1.21 | 0.04 | |||
| BD*(1) C 5 - C 6 | 0.013 | 11.59 | 1.3 | 0.05 | |||
| BD*(1) C 5 - H 11 | 0.012 | 9.46 | 1.16 | 0.05 | |||
| BD*(1) C 6 - H 12 | 0.014 | 4.18 | 1.14 | 0.03 | |||
| BD*(1) C 7 - N 15 | 0.013 | 6.53 | 1.15 | 0.04 | |||
| σ -σ* | BD ( 1) C 1 - N 15 | 1.986 | BD*(1) C 1 - C 2 | 0.027 | 4.44 | 1.36 | 0.03 |
| BD*(1) C 1 - C 6 | 0.021 | 8.54 | 1.38 | 0.05 | |||
| BD*(1) C 2 - C 3 | 0.022 | 9.87 | 1.38 | 0.05 | |||
| BD*(1) C 7 - H 14 | 0.012 | 9.12 | 1.23 | 0.05 | |||
| BD*(1) C 7 - N 15 | 0.013 | 6.4 | 1.26 | 0.04 | |||
| σ -σ* | BD ( 1) C 2 - C 3 | 1.974 | BD*(1) C 1 - C 2 | 0.027 | 15.44 | 1.24 | 0.06 |
| BD*(1) C 1 - N 15 | 0.027 | 7.11 | 1.13 | 0.04 | |||
| BD*(1) C 2 - C 3 | 0.022 | 16.48 | 1.22 | 0.06 | |||
| BD*(1) C 3 - C 4 | 0.013 | 5.1 | 1.25 | 0.04 | |||
| BD*(1) C 7 - C 8 | 0.019 | 12.84 | 1.21 | 0.06 | |||
| BD*(1) C 7 - H 14 | 0.012 | 18.95 | 1.07 | 0.06 | |||
| BD*(1) C 7 - N 15 | 0.013 | 5.44 | 1.09 | 0.03 | |||
| BD*(1) C 8 - C 16 | 0.036 | 10.08 | 1.13 | 0.05 | |||
| BD*(1) C 16 - N 18 | 0.01 | 9.5 | 1.25 | 0.05 | |||
| σ -σ* | BD ( 1) C 3 - C 4 | 1.978 | BD*(1) C 2 - C 3 | 0.023 | 13.77 | 1.27 | 0.06 |
| BD*(1) C 2 - C 8 | 0.027 | 19.25 | 1.19 | 0.07 | |||
| BD*(1) C 3 - H 9 | 0.014 | 4.69 | 1.14 | 0.03 | |||
| BD*(1) C 4 - C 5 | 0.016 | 10.84 | 1.26 | 0.05 | |||
| BD*(1) C 5 - H 11 | 0.012 | 8.49 | 1.15 | 0.04 | |||
| π -π* | BD ( 2) C 3 - C 4 | 1.721 | BD*(2) C 1 - C 2 | 0.477 | 75.1 | 0.28 | 0.07 |
| BD*(2) C 5 - C 6 | 0.319 | 82.34 | 0.28 | 0.07 | |||
| σ -σ* | BD ( 1) C 3 - H 9 | 1.979 | BD*(1) C 1 - C 2 | 0.027 | 17.24 | 1.06 | 0.06 |
| BD*(1) C 4 - C 5 | 0.016 | 16.07 | 1.08 | 0.06 | |||
| σ -σ* | BD ( 1) C 4 - C 5 | 1.979 | BD*(1) C 3 - C 4 | 0.013 | 11.17 | 1.28 | 0.05 |
| BD*(1) C 3 - H 9 | 0.014 | 10.46 | 1.13 | 0.05 | |||
| BD*(1) C 5 - C 6 | 0.013 | 10.92 | 1.27 | 0.05 | |||
| BD*(1) C 6 - H 12 | 0.013 | 11.05 | 1.12 | 0.05 | |||
| σ -σ* | BD ( 1) C 4 - H 10 | 1.98 | BD*(1) C 2 - C 3 | 0.022 | 15.98 | 1.08 | 0.06 |
| BD*(1) C 5 - C 6 | 0.013 | 15.94 | 1.1 | 0.06 | |||
| σ -σ* | BD ( 1) C 5 - C 6 | 1.976 | BD*(1) C 1 - C 6 | 0.021 | 13.97 | 1.27 | 0.06 |
| BD*(1) C 1 - N 15 | 0.026 | 25.98 | 1.14 | 0.08 | |||
| BD*(1) C 4 - C 5 | 0.015 | 10.54 | 1.27 | 0.05 | |||
| BD*(1) C 4 - H 10 | 0.012 | 8.28 | 1.15 | 0.04 | |||
| BD*(1) C 6 - H 12 | 0.013 | 5.27 | 1.13 | 0.03 | |||
| π -π* | BD ( 2) C 5 - C 6 | 1.729 | BD*(2) C 1 - C 2 | 0.477 | 82.01 | 0.28 | 0.07 |
| BD*(2) C 3 - C 4 | 0.301 | 72.63 | 0.29 | 0.06 | |||
| σ -σ* | BD ( 1) C 5 - H 11 | 1.98 | BD*(1) C 1 - C 6 | 0.022 | 14.69 | 1.08 | 0.06 |
| BD*(1) C 3 - C 4 | 0.014 | 15.69 | 1.11 | 0.06 | |||
| σ -σ* | BD ( 1) C 6 - H 12 | 1.98 | BD*(1) C 1 - C 2 | 0.027 | 17.66 | 1.07 | 0.06 |
| BD*(1) C 4 - C 5 | 0.016 | 15.06 | 1.09 | 0.06 | |||
| σ -σ* | BD ( 1) C 7 - C 8 | 1.972 | BD*(1) C 2 - C 3 | 0.023 | 20.42 | 1.28 | 0.07 |
| BD*(1) C 2 - C 8 | 0.027 | 12.18 | 1.21 | 0.05 | |||
| BD*(1) C 7 - H 14 | 0.012 | 6.53 | 1.13 | 0.04 | |||
| BD*(1) C 8 - C 16 | 0.036 | 12.34 | 1.2 | 0.05 | |||
| BD*(1) H 13 - N 15 | 0.017 | 14.02 | 1.09 | 0.05 | |||
| BD*(1) C 16 - H 17 | 0.022 | 4.23 | 1.13 | 0.03 | |||
| π -π* | BD ( 2) C 7 - C 8 | 1.801 | BD*(2) C 1 - C 2 | 0.477 | 66.23 | 0.3 | 0.07 |
| BD*(2) C 7 - C 8 | 0.349 | 8.87 | 0.29 | 0.02 | |||
| BD*(2) C 16 - N 18 | 0.212 | 73.6 | 0.29 | 0.06 | |||
| σ -σ* | BD ( 1) C 7 - H 14 | 1.984 | BD*(1) C 1 - N 15 | 0.026 | 12.26 | 1.01 | 0.05 |
| BD*(1) C 2 - C 8 | 0.027 | 8.66 | 1.06 | 0.04 | |||
| BD*(1) C 7 - C 8 | 0.019 | 6.07 | 1.13 | 0.04 | |||
| σ -σ* | BD ( 1) C 7 - N 15 | 1.985 | BD*(1) C 1 - C 6 | 0.021 | 17.03 | 1.39 | 0.07 |
| BD*(1) C 1 - N 15 | 0.026 | 7.32 | 1.26 | 0.04 | |||
| BD*(1) C 7 - C 8 | 0.019 | 5.06 | 1.38 | 0.04 | |||
| BD*(1) C 8 - C 16 | 0.036 | 16.07 | 1.3 | 0.06 | |||
| σ -σ* | BD ( 1) C 8 - C 16 | 1.979 | BD*(1) C 1 - C 2 | 0.027 | 4.81 | 1.23 | 0.03 |
| BD*(1) C 2 - C 8 | 0.027 | 14.31 | 1.18 | 0.06 | |||
| BD*(1) C 7 - C 8 | 0.019 | 14.81 | 1.24 | 0.06 | |||
| BD*(1) C 7 - N 15 | 0.013 | 4.48 | 1.13 | 0.03 | |||
| BD*(1) C 16 - N 18 | 0.01 | 9.54 | 1.29 | 0.05 | |||
| σ -σ* | BD ( 1) H 13 - N 15 | 1.99 | BD*(1) C 1 - C 2 | 0.027 | 7.41 | 1.24 | 0.04 |
| BD*(1) C 7 - C 8 | 0.019 | 5.73 | 1.26 | 0.04 | |||
| σ -σ* | BD ( 1) C 16 - H 17 | 1.969 | BD*(1) C 7 - C 8 | 0.019 | 20.08 | 1.07 | 0.06 |
| BD*(1) N 18 - N 19 | 0.03 | 35.31 | 0.89 | 0.08 | |||
| σ -σ* | BD ( 1) C 16 - N 18 | 1.988 | BD*(1) C 2 - C 8 | 0.027 | 4.98 | 1.39 | 0.04 |
| BD*(1) C 8 - C 16 | 0.036 | 9.37 | 1.38 | 0.05 | |||
| BD*(1) N 19 - C 21 | 0.073 | 10.33 | 1.33 | 0.05 | |||
| π -π* | BD ( 2) C 16 - N 18 | 1.942 | BD*(2) C 7 - C 8 | 0.349 | 32.55 | 0.35 | 0.05 |
| σ -σ* | BD ( 1) N 18 - N 19 | 1.986 | BD*(1) C 16 - H 17 | 0.022 | 8.62 | 1.27 | 0.05 |
| BD*(1) C 21 - O 29 | 0.021 | 5.69 | 1.44 | 0.04 | |||
| σ -σ* | BD ( 1) N 19 - H 20 | 1.983 | BD*(1) C 21 - C 22 | 0.066 | 15.1 | 1.09 | 0.06 |
| σ -σ* | BD ( 1) N 19 - C 21 | 1.99 | BD*(1) C 16 - N 18 | 0.01 | 8.91 | 1.42 | 0.05 |
| BD*(1) C 22 - C 23 | 0.021 | 4.39 | 1.39 | 0.03 | |||
| BD*(1) C 25 - C 23 | 0.026 | 4.23 | 4.67 | 0.06 | |||
| σ -σ* | BD ( 1) C 21 - C 22 | 1.974 | BD*(1) N 19 - H 20 | 0.042 | 10.96 | 1.03 | 0.05 |
| BD*(1) C 21 - O 29 | 0.021 | 5.27 | 1.24 | 0.04 | |||
| BD*(1) C 22 - C 23 | 0.021 | 7.07 | 1.23 | 0.04 | |||
| BD*(1) C 22 - C 24 | 0.033 | 8.12 | 1.22 | 0.04 | |||
| BD*(1) C 23 - C 25 | 0.015 | 9.08 | 1.25 | 0.05 | |||
| BD*(1) C 25 - N 32 | 0.015 | 9.5 | 1.21 | 0.05 | |||
| σ -σ* | BD ( 1) C 21 - O 29 | 1.992 | BD*(1) N 18 - N 19 | 0.03 | 7.28 | 1.39 | 0.04 |
| BD*(1) C 21 - C 22 | 0.066 | 7.11 | 1.45 | 0.05 | |||
| BD*(1) C 22 - C 24 | 0.033 | 5.61 | 1.59 | 0.04 | |||
| π -π* | BD ( 2) C 21 - O 29 | 1.979 | BD*(2) C 22 - C 24 | 0.328 | 13.85 | 0.42 | 0.04 |
| σ -σ* | BD ( 1) C 22 - C 23 | 1.973 | BD*(1) N 19 - C 21 | 0.073 | 8.49 | 1.13 | 0.04 |
| BD*(1) C 21 - C 22 | 0.066 | 7.07 | 1.12 | 0.04 | |||
| BD*(1) C 22 - C 24 | 0.033 | 14.85 | 1.26 | 0.06 | |||
| BD*(1) C 23 - C 25 | 0.0148 | 10.84 | 1.29 | 0.05 | |||
| BD*(1) C 24 - H 27 | 0.023 | 9.5 | 1.15 | 0.05 | |||
| BD*(1) C 25 - H 28 | 0.013 | 10.59 | 1.14 | 0.05 | |||
| σ -σ* | BD ( 1) C 22 - C 24 | 1.979 | BD*(1) C 21 - C 22 | 0.066 | 6.23 | 1.13 | 0.04 |
| BD*(1) C 21 - O 29 | 0.021 | 6.65 | 1.29 | 0.04 | |||
| BD*(1) C 22 - C 23 | 0.021 | 16.07 | 1.27 | 0.06 | |||
| BD*(1) C 23 - H 26 | 0.014 | 10.42 | 1.16 | 0.05 | |||
| BD*(1) C 24 - H 27 | 0.023 | 4.35 | 1.15 | 0.03 | |||
| BD*(1) C 24 - C 30 | 0.015 | 5.9 | 1.26 | 0.04 | |||
| π -π* | BD ( 2) C 22 - C 24 | 1.61 | BD*(2) C 21 - O 29 | 0.274 | 74.39 | 0.29 | 0.07 |
| BD*(2) C 23 - C 25 | 0.287 | 97.53 | 0.28 | 0.07 | |||
| BD*(2) C 28 - N 31 | 0.372 | 68.03 | 0.27 | 0.06 | |||
| σ -σ* | BD ( 1) C 23 - C 25 | 1.979 | BD*(1) C 21 - C 22 | 0.066 | 12.01 | 1.13 | 0.05 |
| BD*(1) C 22 - C 23 | 0.021 | 12.22 | 1.27 | 0.05 | |||
| BD*(1) C 25 - H 28 | 0.014 | 4.35 | 1.15 | 0.03 | |||
| BD*(1) C 30 - H 31 | 0.024 | 9.25 | 1.14 | 0.05 | |||
| π -π* | BD ( 2) C 23 - C 25 | 1.639 | BD*(2) C 22 - C 24 | 0.329 | 69.33 | 0.29 | 0.06 |
| BD*(2) C 30 - N 32 | 0.373 | 123.22 | 0.27 | 0.08 | |||
| σ -σ* | BD ( 1) C 23 - H 26 | 1.979 | BD*(1) C 22 - C 24 | 0.033 | 16.99 | 1.08 | 0.06 |
| σ -σ* | BD ( 1) C 24 - H 27 | 1.979 | BD*(1) C 22 - C 23 | 0.021 | 17.45 | 1.08 | 0.06 |
| BD*(1) C 30 - N 32 | 0.016 | 20.33 | 1.06 | 0.06 | |||
| σ -σ* | BD ( 1) C 24 - N 31 | 1.986 | BD*(1) C 21 - C 22 | 0.066 | 9.41 | 1.25 | 0.05 |
| BD*(1) C 22 - C 24 | 0.033 | 8.12 | 1.38 | 0.05 | |||
| BD*(1) C 28 - H 30 | 0.024 | 8.74 | 1.25 | 0.05 | |||
| σ -σ* | BD ( 1) C 25 - C 23 | 1.985 | BD*(1) C 23 - C 25 | 0.015 | 10.59 | 1.3 | 0.05 |
| BD*(1) C 23 - H 26 | 0.014 | 11.63 | 1.16 | 0.05 | |||
| BD*(1) C 30 - N 32 | 0.016 | 5.86 | 1.26 | 0.04 | |||
| σ -σ* | BD ( 1) C 25 - H 23 | 1.979 | BD*(1) C 22 - C 23 | 0.021 | 14.52 | 1.09 | 0.06 |
| BD*(1) C 30 - N 32 | 0.016 | 17.87 | 1.07 | 0.06 | |||
| σ -σ* | BD ( 1) C 30 - H 31 | 1.982 | BD*(1) C 23 - C 25 | 0.015 | 14.73 | 1.11 | 0.06 |
| BD*(1) C 25 - N 32 | 0.015 | 20.17 | 1.07 | 0.06 | |||
| σ -σ* | BD ( 1) C 30 - N 32 | 1.987 | BD*(1) C 24 - H 27 | 0.023 | 8.79 | 1.27 | 0.05 |
| BD*(1) C 25 - H 29 | 0.013 | 6.11 | 1.26 | 0.04 | |||
| π -π* | BD ( 2) C 30 - N 32 | 1.705 | BD*(2) C 22 - C 24 | 0.328 | 116.4 | 0.32 | 0.09 |
| BD*(2) C 23 - C 25 | 0.287 | 52.38 | 0.32 | 0.06 | |||
| n -π* | LP ( 1) N 15 | 1.613 | BD*(2) C 1 - C 2 | 0.476 | 140.71 | 0.3 | 0.09 |
| BD*(2) C 7 - C 8 | 0.349 | 160.41 | 0.29 | 0.1 | |||
| n -σ* | LP ( 1) N 18 | 1.921 | BD*(1) C 8 - C 16 | 0.036 | 42.76 | 0.87 | 0.09 |
| BD*(1) C 16 - H 17 | 0.022 | 16.74 | 0.8 | 0.05 | |||
| BD*(1) N 19 - H 20 | 0.042 | 33.05 | 0.75 | 0.07 | |||
| n -π* | LP ( 1) N 19 | 1.679 | BD*(2) C 16 - N 18 | 0.212 | 102.42 | 0.3 | 0.08 |
| BD*(1) C 21 - O 29 | 0.021 | 5.4 | 0.87 | 0.03 | |||
| BD*(2) C 21 - O 29 | 0.274 | 170.41 | 0.33 | 0.1 | |||
| n -σ* | LP ( 1) N 32 | 1.917 | BD*(1) C 22 - C 24 | 0.033 | 39.46 | 0.89 | 0.08 |
| BD*(1) C 24 - H 27 | 0.023 | 16.57 | 0.78 | 0.05 | |||
| BD*(1) C 25 - C 23 | 0.026 | 8.16 | 4.18 | 0.08 | |||
| BD*(1) C 30 - H 31 | 0.023 | 17.11 | 0.77 | 0.05 | |||
| n -σ* | LP ( 1) O 29 | 1.979 | BD*(1) C 21 - C 22 | 0.066 | 8.24 | 1.12 | 0.04 |
| n -σ* | LP ( 2) O 29 | 1.868 | BD*(1) N 19 - C 21 | 0.073 | 99.66 | 0.68 | 0.12 |
| BD*(1) C 21 - C 22 | 0.066 | 73.35 | 0.68 | 0.1 | |||
| BD*(1) C 25 - C 23 | 0.026 | 6.9 | 4.1 | 0.08 | |||
| π*-π* | BD*( 2) C 1 - C 2 | 0.477 | BD*(2) C 3 - C 4 | 0.301 | 1195.8 | 0.01 | 0.08 |
| π*-π* | BD*( 2) C 7 - C 8 | 0.349 | BD*(2) C 1 - C 2 | 0.477 | 711.11 | 0.01 | 0.06 |
| π*-σ* | BD*( 2) C 21 - O 29 | 0.274 | BD*(1) C 21 - O 29 | 0.021 | 18.24 | 0.54 | 0.11 |
| π*-π* | BD*( 2) C 30 - N 32 | 0.373 | BD*(2) C 22 - C 24 | 0.328 | 604.96 | 0.02 | 0.08 |
| BD*(2) C 23 - C 25 | 0.287 | 714.04 | 0.02 | 0.08 |
HOMO–LUMO Analysis
The HOMO–LUMO plot of ICINH molecule is shown in Figure 4 In HOMO diagram, the colored portions indicate the prominent donor levels which contribute in the electronic transitions and similarly the LUMO diagram indicates the prominent acceptors level through colored shades which involve in the electronic transitions. Homo localized in the indole and carbonyl group. The LUMO is located over the pyridine and hydrazone linkage. The molecule ICINH has lower energy gap and hence the probability of π-π* proton transition is highly possible in between HOMO and LUMO orbitals. The HOMO/LUMO energies are calculated using B3LYP/6-311++G(d,p) level.
Figure 4.The frontier molecular orbitals of ICINH
By using HOMO and LUMO energy value ICINH, the global chemical reactivity descriptors such as hardness (η), chemical potential (μ), softness (S), electronegativity (χ) and electrophilicity index (ω) have been calculated and are listed in Table 5. It can be expressed through HOMO and LUMO orbital energies as I=-EHOMO and A=-ELOMO, the electron affinity I and Ionization potential A of title molecule ICINH are also calculated by using B3LYP/6-311++G(d,p) basis set. The calculated values of the softness, hardness, chemical potential electronegativity and electrophilicity index, Homo, Lumo and energy gap of the molecule are: 4.341 eV, 3.954 eV, 3.954 eV and 1.801 eV, -6.125 eV, -1.784 eV and 4.341 eV, respectively. The soft molecule has small energy gap and hard molecule has large energy gap. In addition, the frontier molecular orbital energies are also calculated using the same basis set and are listed in Table 6.
Table 5. The Physico-chemical properties of ICINH| Parameters | Values |
| HOMO | -6.125 eV |
| LUMO | -1.784 eV |
| Energy gap | 4.341 eV |
| Ionization potential (IP) | 6.125 eV |
| Electron affinity (EA) | 1.784 eV |
| Electrophilicity Index (ω) | 1.801 |
| Chemical Potential (µ) | 3.954 |
| Electronegativity (χ) | -3.954 |
| Hardness (η) | -4.341 |
| Softness (S) | 8.682 |
| Occupancy | Orbital energies(a.u) | Orbital energies(eV) | Kinetic energies(a.u) |
| O52 | -0.269 | -7.319 | 1.267 |
| O53 | -0.267 | -7.265 | 1.605 |
| O54 | -0.262 | -7.129 | 1.871 |
| O55 | -0.254 | -6. 911 | 1.250 |
| O56 | -0.228 | -6.203 | 1.576 |
| V57 | -0.071 | -1.931 | 1.664 |
| V58 | -0.055 | -1.496 | 1.519 |
| V59 | -0.034 | 0. 925 | 1.367 |
| V60 | 0.025 | 0.680 | 0.381 |
| V61 | 0.023 | 0.625 | 1.250 |
UV-Vis Spectra
The UV absorption spectrum for ICINH is recorded in the range 200-800 nm. All the structures allow strong π-π* and σ-σ* transition in the UV-Vis region with high extinction coefficients. The calculated results involving in the vertical excitation energies, oscillator strength (f) and wavelength are carried out and compare with measured experimental wavelength. Typically, according to Frank-Condon principle, the maximum absorption peaks (λmax) in a UV-Vis spectrum correspond to vertical excitation. The λmax of ICINH molecule are calculated using TD-DFT/6-311++G(d,p) basis set. It is evident from the Table 7, the possible π-π* transitions, with absorption maximum at 413.14, 389.2, 357.32 nm, belong to gas phase and the oscillator strength for respective transitions are 0.0009, 0.0136 and 0.0296, respectively. The calculated absorption maxima has been found to be 357.32 nm which is moderately coincides with the experimental value 342.42 nm. The combined theoretical and experimental UV-Vis absorption spectra are shown in Figure 5.
Table 7. The electronic transition of ICINH| Calculated at B3LYP/6-311++G(d,p) | Oscillator strength | CalculatedBand gap (ev/nm) | ExperimentalBand gap (ev/nm) | Type |
| Excited State-1 | Singlet-A (f=0.0009) | 3.0010 eV/413.14 nm | ||
| 69 -> 70 | 0.6908 | |||
| Excited State-2 | Singlet-A (f=0.0136) | 3.1849 eV/389.29 nm | ||
| 66 -> 70 | -0.1059 | |||
| 68 -> 70 | 0.6587 | |||
| 68 -> 71 | 0.1308 | |||
| Excited State-3 | Singlet-A (f=0.0296) | 3.4698 eV/357.32 nm | 342.42 | π-π* |
| 66 -> 70 | 0.1243 | |||
| 66 -> 71 | 0.1564 | |||
| 68 -> 70 | -0.1275 | |||
| 68 -> 71 | 0.4689 | |||
| 69 -> 71 | -0.4067 |
Figure 5.The combined theoretical and experimental UV-Visible spectra of ICINH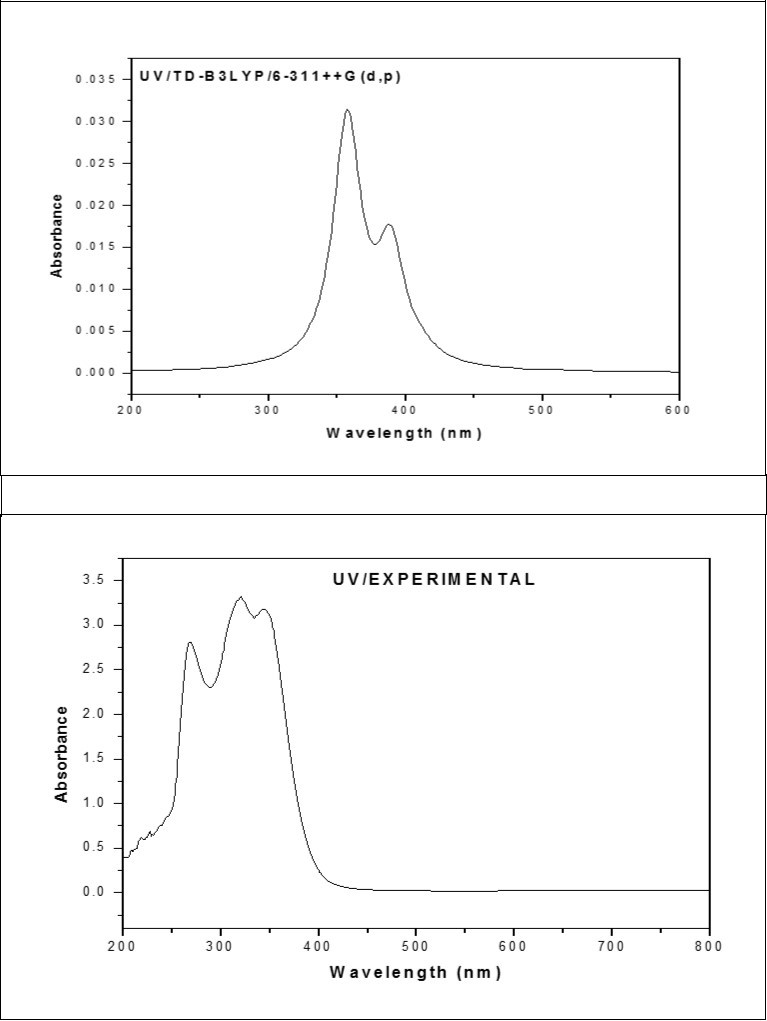
Molecular Electrostatic Potential
In the present study, MEP surface map of ICINH is calculated using B3LYP/6-311++G(d,p) basis set and illustrated in Figure 6 The MEP which is a plot of electrostatic potential map onto the constant ED surface. In the majority of the MEPs, the maximum negative region which is the preferred site for electrophilic attack and the maximum positive region is the preferred site for nucleophilic attack. The importance of MEPs lies in the fact that it simultaneously displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions in terms of colour scheme (Figure 6) and is very useful in research of molecular structure with its physiochemical property relationship 26, 27. The color scheme for the MEP surface is as follows: red for electron rich, partially negative charge: blue correspond to electron deficient, partially positive charge: green for neutral, respectively 28, 29. The electrostatic potential increases in the order of red<orange<yellow<green<blue. The color code of the map range between -9.185 e-2 (deepest red) to 9.185 e-2 (deepest blue). It is seen from the MEP map of the title molecule, the regions having the negative potential over the electronegative atom (oxygen atom), and the regions having the positive potential over all the hydrogen atoms in indole ring. The oxygen atom indicates the strong repulsion with other atom. These two ends of the molecule which are positively and negatively charged are prone to electrophilic and electrophobic reactions with other molecules.
Figure 6.The molecular electrostatic potential map of ICINH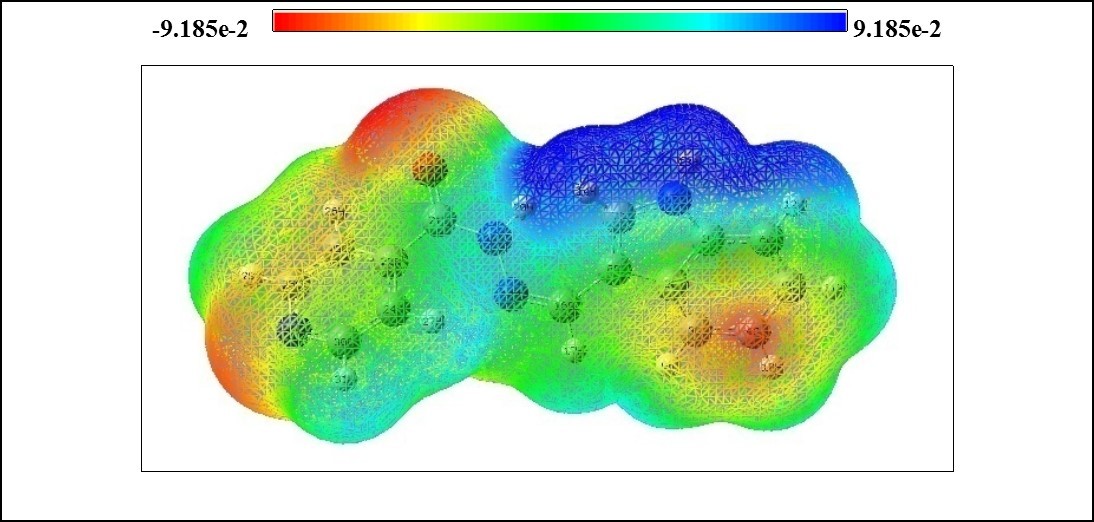
Mulliken Atomic Charges
It is well known that the atomic charges are very much dependent on how the atoms are defined. It also plays an important role in the application of quantum chemical calculation to molecular system because of atomic charges affect the dipole moment, molecular polarizability, electronic structure and a lot of properties of molecular systems. The mulliken charges calculated at B3LYP/6-311++G(d,p) basis set for the molecule under study are given in Table 8. The mulliken atomic charges plot for ICINH is shown in Figure 7
The C2 atom has high positive charge which is due to the indole group. Similarly the C16 atom has high negative charge due to the attachment of C16=N18 group. All the hydrogen atoms have positive charge.
Table 8. The Mulliken atomic charges of ICINH| Atoms | Charges | Atoms | Charges |
| 1C | -0.2987 | 17H | 0.1624 |
| 2C | 1.3144 | 18N | 0.0896 |
| 3C | -0.6692 | 19N | -0.0922 |
| 4C | -0.5107 | 20H | 0.2843 |
| 5C | -0.0917 | 21C | -0.5353 |
| 6C | -0.6358 | 22C | 0.7243 |
| 7C | -0.2138 | 23C | 0.0949 |
| 8C | 0.8639 | 24C | -0.5605 |
| 9H | 0.1424 | 25C | -0.1468 |
| 10H | 0.1605 | 26H | 0.2244 |
| 11H | 0.1674 | 27H | 0.2180 |
| 12H | 0.1439 | 28C | -0.2971 |
| 13H | 0.3039 | 29H | 0.1812 |
| 14H | 0.2100 | 30H | 0.1860 |
| 15N | -0.0449 | 31N | -0.0054 |
| 16C | -1.1123 | 32O | -0.2591 |
Figure 7.The Mulliken atomic charges of ICINH
Thermodynamic Properties
On the basis of vibrational analysis, the standard statistical thermodynamic functions: heat capacity (C), entropy (S) and enthalpy changes (H) for the title molecule are obtained from the theoretical harmonic frequencies and are listed in Table 9. From Table 10, it can be observed that these thermodynamic functions are increasing with temperature ranging from 100 to 1000 K. The obvious reason for this is almost linear increase, and this is due to the increase in internal energy of the molecule in accordance with kinetic theory of gases due to the fact that the molecular vibrational intensities increase with temperature 30. The correlation equations between heat capacities, entropies, enthalpy, changes and temperatures are fitted by quadratic formulas and the corresponding fitting factors (R2) for these thermodynamic properties are 0.99905, 0.9994 and 0.99998, respectively and the correlation graphics are shown in Figure 8.
Table 9. The calculated total energy (a.u), zero point vibrational energies (Kcal/mol), rotational constants (GHz) and entropy (cal/mol K-1) for ICINH| Parameters | B3LYP/6-311++G(d,p) |
| Total Energies | -873.259 |
| Zero-point Energy | 154. 930 (Kcal/Mol) |
| Rotational constants (GHZ) | 1.119 |
| 0.128 | |
| 0.120 | |
| Entropy | |
| Total | 131.409 |
| Translational | 42.613 |
| Rotational | 34.185 |
| Vibrational | 54.610 |
| T (K) | S (J/mol.K) | Cp (J/mol.K) | ddH (kJ/mol) |
| 100.00 | 362.62 | 109.42 | 7.39 |
| 200.00 | 460.26 | 183.91 | 21.90 |
| 298.15 | 549.35 | 268.44 | 44.07 |
| 300.00 | 551.01 | 270.03 | 44.57 |
| 400.00 | 640.10 | 351.39 | 75.73 |
| 500.00 | 726.10 | 419.62 | 114.40 |
| 600.00 | 807.63 | 474.29 | 159.20 |
| 700.00 | 884.14 | 517.90 | 208.89 |
| 800.00 | 955.68 | 553.15 | 262.50 |
| 900.00 | 1022.55 | 582.09 | 319.31 |
| 1000.00 | 1085.17 | 606.19 | 378.76 |
Figure 8.The thermodynamic properties of ICINH at different temperatures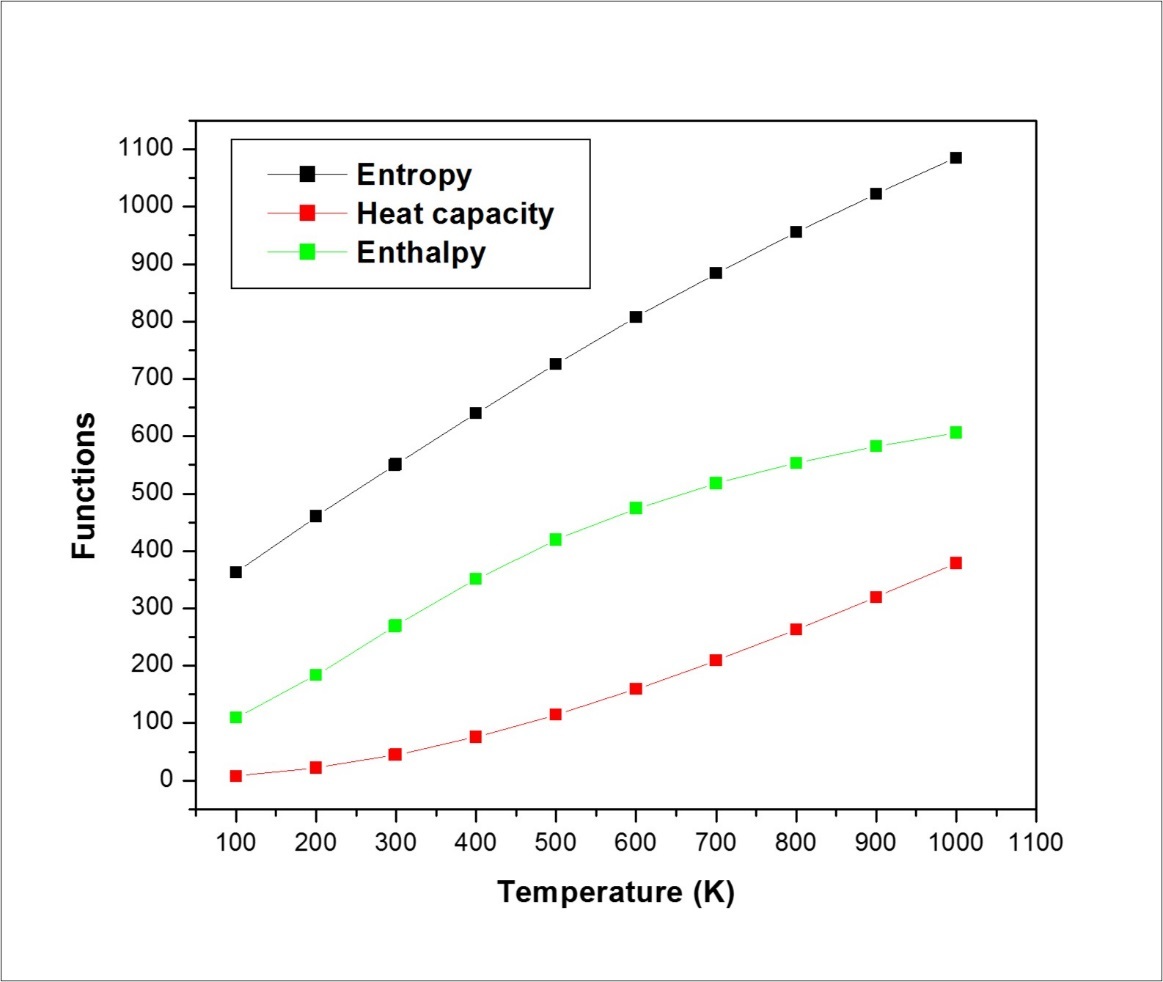
They can be used to compute the other thermodynamic energies according to relationships of thermodynamic functions and estimate directions of chemical reactions according to the second law of thermodynamics in thermo chemical field 31. In this study all thermodynamic calculations are done in gas phase and they could not be used in solution.
C0p,m = 6.76926 + 0.02858T + 2.52495x10-5 T2 (R2 = 0.99905)
S0m = 1.40875 + 0.00595T + 5.25468x10-5 T2 (R2 = 0.99998)
ΔH0m = 4.06057 + 0.01714T + 1.51461x10-5 T2 (R2 = 0.9994)
Conclusion
The FT-IR, FT-Raman and UV-Vis spectra of the compound ICINH had been recorded and analyzed. The detailed interpretations of the vibrational spectra had been carried out. The optimized geometrical parameters were calculated and compared with the reported XRD data. The vibrational assignments were further justified with help of the PED analysis. The HOMO-LUMO energy gap indicated the stability and reactivity of the title compound. The good correlation between the UV-Vis, absorption maxima and calculated electronic absorption maxima were found. The donor-acceptor interaction, as obtained from NBO analysis could fairly explains the decrease of occupancies of σ bonding orbital and the increase of occupancy of π* anti-bonding orbitals. In addition, mulliken atomic charges, MEP, Thermodynamic parameters, first order hyperpolarizabilities and dipole moment of the title compound were also calculated.
References
- 3.Billes F, Podea P V, Mohammed-Ziegler I, Tosa M, Mikosch H et al. (2009) . , Spectrochim. Acta A 74, 1031-1045.
- 6.Vasantha Kumar V, Gouda S L, J Laxmikanth Rao. (2013) Ab initioand density functional theory studies on vibrational spectra of 3-hydroxy-3-(2-methyl-1H-indol-3-yl)indolin-2-one, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 103. 304-310.
- 7.Saleem H, Subashchandrabose S, Erdogduc Y, Thanikachalam V, Jayabharathi J. (2013) FT-IR, FT-Raman spectral and conformational studies on (E)-2-(2-hydroxybenzylidenamino)-3-(1H-indol-3yl) propionic acid, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 101, 91-99.
- 8.N Ramesh Babu, Subashchandrabose S, Padusha M Syed Ali, Saleem H, Manivannan V et al. (2014) Synthesis and structural characterization of (E)-N0-((Pyridin-2-yl)methylene) benzohydrazide by X-ray diffraction, FT-IR, FT-Raman and DFT methods. , Journal of Molecular Structure 1072, 84-93.
- 9.Lissoni P, Bucovec R, Bonfonti A, Giani L, Mandelli A et al. (2001) Density Functional Theory of Atoms and molecules. , Oxford, New York,1989 30(2), 123-15.
- 13.James C, Pettit G R, Nielsen O F, Jayakumar V S, Joe T H. (2008) . , Spectrochimm. Acta A 70, 1208-1216.
- 14.Dickson Babu, Gachumale S, Sambandam A, AirodyVasudeva A. (2015) New D-p-A Type indole based chromoogens for DSSC: Design Synthesis and Performance studies’, Dyes and Pig. 112, 183-191.
- 15.Hassan M, Former W, Badawi.analysis of the molecular Stutucture and vibrational spectra of the title indole based analgesic drug indomethacin”. , Spectrochim. Acta A: Mol. Biomol. Spectrosc 123, 447-454.
- 17.Rauhut G, Pulay P. (1995) Transferable Scaling Factors for Density Functional Derived Vibrational Force Fields. , J.Phys.Chem 99, 3093-3100.
- 22.Silverstein M, G Clayton Basseler, Morill C. (1981) Spectrometric Identification of Organic Compounds,Wiley,New. , York
- 24.Nikitina A N, Yakovlev L P, Fedotov N S, Mikhailov B M. (1967) . , Zhurnal Prikladnoi spektroskopii 6(2), 232-238.
- 26.Murray J S, Sen K. (1996) Molecular Electrostatic Potentials, Concepts and Applications,Elsevier,Amsterdam.
- 27.Scrocco E, Tomasi J. (1978) . In:P.Lowdin(Ed.) Advances in Quantum Chemistry,Academic , Press,New York .